
Figure 1. A screenshot of the Nature Communications paper.
Synthetic lethality (SL), a genetic interaction where simultaneous mutations or perturbations in two genes lead to cell death whereas mutation of a single gene does not produce this effect, has emerged as a promising strategy for cancer treatment strategy. By targeting SL partner genes of genes with cancer-specific mutations, researchers can kill cancer cells without harming healthy cells. Although SL has been known for over a century, its practical application still faces challenges, particularly in the rapid and accurate identification of SL gene pairs in cancer.
To expedite experimental screening and reduce its cost, researchers have increasingly employed machine learning techniques in recent years to predict SL gene pairs. These methods leverage vast biological datasets to efficiently identify potential SL interactions, narrowing the scope for experimental validation. However, despite the employment of various machine learning models and algorithms, a systematic evaluation of their performance across different scenarios was lacking, making it challenging for researchers to select the appropriate AI tools for SL prediction.
A recent study led by Associate Professor Zheng Jie from the School of Information Science and Technology (SIST) at ShanghaiTech University, in collaboration with other researchers, has filled this critical gap. Published in a paper titled “Benchmarking Machine Learning Methods for Synthetic Lethality Prediction in Cancer” in Nature Communications, the study systematically evaluated 12 state-of-the-art (SOTA) machine learning methods, covering a range of algorithms from traditional machine learning to deep learning, in predicting synthetic lethality (SL) as anticancer drug targets across multiple evaluation scenarios. This research provides scientists with a detailed guide to selecting the most suitable SL prediction tools, thereby advancing the development of precision anticancer therapeutics. Notably, 10 of the 12 machine learning methods evaluated in the study were developed by Zheng Jie and Wu Min from A*STAR in Singapore, the paper’s corresponding authors, whose prior work has set the foundation for many recent advancements in SL prediction.
The research team compiled and established a benchmark dataset and designed various experimental scenarios, including three data partitioning methods, four positive-negative sample ratios, and three negative sample selection methods, to assess the ability of these models to classify and rank candidate SL gene pairs across different scenario combinations (Figure 2). By comparing these models, the research team found that improvement of data quality, such as selecting negative samples with higher quality, significantly enhanced the performance of all methods.
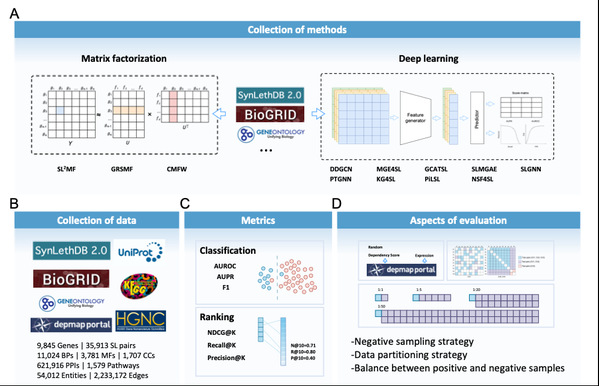
Figure 2. The collection of methods, sources of benchmark data, and evaluation setting.
Among all the evaluated methods, the SLMGAE (SL prediction with multi-view graph auto-encoder) algorithm performed the best overall. However, these methods still showed certain limitations in handling real-world applications. Notably, in the “cold-start” test—where models were tested on a new list of gene pairs of which neither gene had been involved in training—the performance of all the methods decreased. Additionally, for specific cancer contexts (e.g., prediction of cell-line specific SL), the models’ predictive accuracy and generalization capabilities need further improvement.
The results from this benchmarking study have provided scientists with a practical guide for selecting SL prediction methods and revealed the limitations of current machine learning methods in practical applications. This study offers valuable references for future studies aiming to develop more accurate and reliable SL prediction tools, thus providing researchers in the field of SL and anticancer drug discovery with important digital resources and insights to accelerate targeted therapies for cancer.
Third-year PhD student Feng Yimiao from Zheng’s group is the first author of the paper. Contributors to this project include: Long Yahui, scientist from the Bioinformatics Institute, A*STAR, Singapore; Li Quan, assistant professor from SIST; and master’s students Wang He and Ouyang Yang. Graduate students Mao Weifan, Yue Zhen, Tao Siyu, and Yang Yang from SIST provided support for the project. The work was initiated as a project of the SIST course CS286 (AI for Science and Engineering). The paper’s corresponding authors are Wu Min, principal scientist at Institute for Infocomm Research, A*STAR, and Zheng Jie. ShanghaiTech University is the primary affiliation.
*This article is provided by Prof. Zheng Jie