近日,上海科技大学生命科学与技术学院张力烨课题组在生物信息学期刊《生物信息学简报》(Briefings in Bioinformatics)发表了题为“Benchmarking copy number aberrations inference tools using single-cell multi-omics datasets”的研究论文,评估了从基于单细胞测序数据推断拷贝数变异的五种广泛使用的生物信息学工具,填补了该领域的关键空白,为研究人员选择和使用拷贝数变异(Copy number aberration,CNA)推断工具提供了指导。
CNA是一种重要的基因组变异类型,在癌症的发生和发展中起着至关重要的作用。肿瘤细胞中CNA的鉴定对早期肿瘤检测、描述肿瘤异质性、了解肿瘤进展和揭示治疗耐药机制具有重要意义。单细胞DNA测序(scDNA-seq)非常适合在单细胞分辨率下鉴定CNA。scDNA-seq虽然能够提供单细胞分辨率的CNA检测能力,但因其技术的局限性——包括测序覆盖度不足与实验成本过高,严重制约了该技术在拷贝数变异研究中的规模化应用。为此,计算生物学专家们已开发出多种基于scRNA-seq数据的CNA推断算法,通过利用基因表达量与拷贝数之间的相关性,成功实现了肿瘤内异质性的高分辨率解析。但目前尚未建立系统化的评估框架,这些计算方法在真实临床场景中的准确性、稳健性与可重复性仍缺乏跨平台验证。
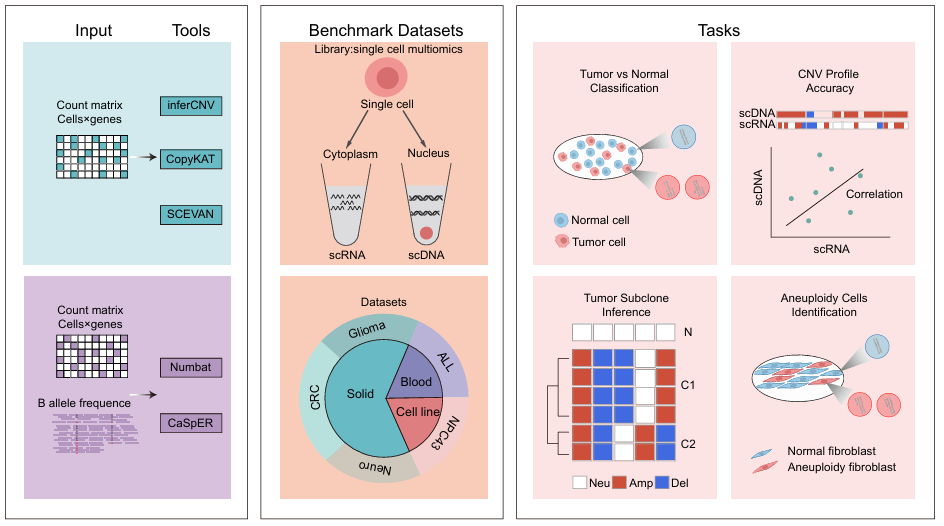
图1:benchmark工作流程
本研究创新性地采用了单细胞同时采集DNA-Seq和RNA-Seq的已有公共数据集,通过使用从scDNA-seq中识别出的拷贝数变异作为评估的金标准,评估了从scRNA-seq中多个维度推断CNA工具的准确性,并论证了参考细胞设置、肿瘤微环境(TME)细胞的包含、肿瘤类型和肿瘤纯度对CNA推断的影响。此外,该研究还评估了肿瘤细胞和正常细胞分类的准确性、CNA推断的准确性、肿瘤亚克隆推断以及非恶性细胞的整倍体识别。
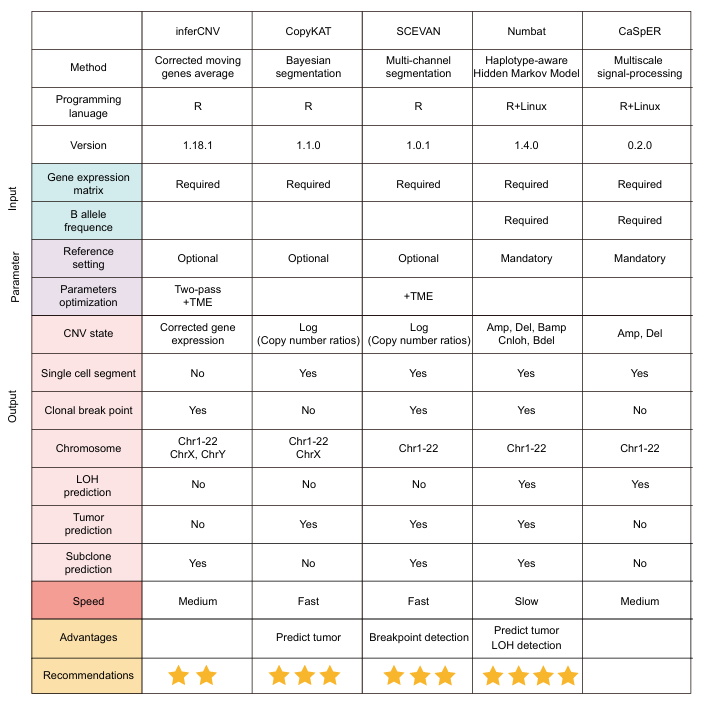
图2:所有方法的评估结果
本研究观察到,所有五种工具均能在一定程度上区分肿瘤细胞和正常细胞,并能较为准确地推断拷贝数变异图谱。在这些方法中,Numbat在各项评估标准中表现最佳。对于仅有表达矩阵可用的情况,推荐使用CopyKAT作为首选方法。同时也展示了目前方法的一些局限,提示一些未来方法学的开发和改进的需求。整体的评估结果(图3)为临床研究人员提供了客观的选型依据。
张力烨教授为本文独立通讯作者。之江实验室宋敏芳(上海科技大学博士毕业生),张力烨课题组硕士研究生马帅为本文共同第一作者。上海科技大学超算中心为多组学数据产出和生物信息分析提供了大力支持。
 沪公网安备 31011502006855号
沪公网安备 31011502006855号