多梳抑制复合物2(PRC2)通过催化亚基EZH2介导H3K27me3修饰,在转录抑制、染色质压缩及胚胎发育调控中发挥关键作用,其功能异常与多种癌症的发生发展密切相关。上海科技大学生命学院戚炜实验室长期研究表观遗传蛋白机器PRC2在肿瘤及其他重大疾病中的病理机制与创新药物研发,前期已阐明PRC2抑制剂在肿瘤中发挥作用的药理机制。近日,戚炜团队与合作者在《先进科学》(Advanced Science)在线发表最新研究,报道了一种EZH2高选择性低毒性小分子变构抑制剂C36的作用机制。
EZH2蛋白被视为极具潜力的干预靶点,但早期进入临床的EZH2抑制剂普遍受困于两大技术瓶颈。一是对高度同源的EZH1蛋白缺乏分辨能力,被认为是诱发血小板减少及贫血等血液学不良反应的主要诱因;二是作用路径过度集中于竞争性占据辅因子SAM的底物结合位点或EED亚基的识别口袋,增加了获得性耐药的发生几率。

全新别构位点,选择性突破440倍
针对现有抑制剂的痛点,研究团队设计筛选出全新别构调节分子C36。体外酶学实验显示,C36对EZH2/PRC2复合物半数抑制浓度(IC50)达到2.27 nM,而对EZH1的活性则几乎无影响(IC50 > 1000 nM),其选择性超过440倍,显著优于目前公开报道的同类化合物。
更关键的是作用机制的不同:C36既不与SAM或底物直接竞争,也不作用于EED亚基的组蛋白H3K27me3结合口袋,而是靶向EZH2蛋白上由SAL结构域、SRM基序及I-SET结构域共同构成的别构位点,通过诱导蛋白质构象的重排,阻断了PRC2复合物内部亚基间的变构通讯。这种新的干预方式赋予了C36极高的细胞层面特异性——在G401肾癌细胞中,C36剂量依赖性地削减了全局H3K27me3的修饰,但对H3K27me1、H3K27me2以及其它赖氨酸位点的甲基化状态几乎不产生扰动;在携带EZH2野生型或获得性突变体的弥漫大B细胞淋巴瘤(DLBCL)系中,该化合物同样实现了对H3K27me3的有效抑制,并成功解除下游靶基因的转录沉默。通过构建基于蛋白降解的EZH2功能缺失模型验证发现,尽管EZH1仍能维持H3K27me1/2水平,但C36几乎不干扰这些修饰,而EED抑制剂和SAM竞争性抑制剂却可有效抑制EZH1相关活性,这从反向角度进一步佐证了C36的卓越选择性。
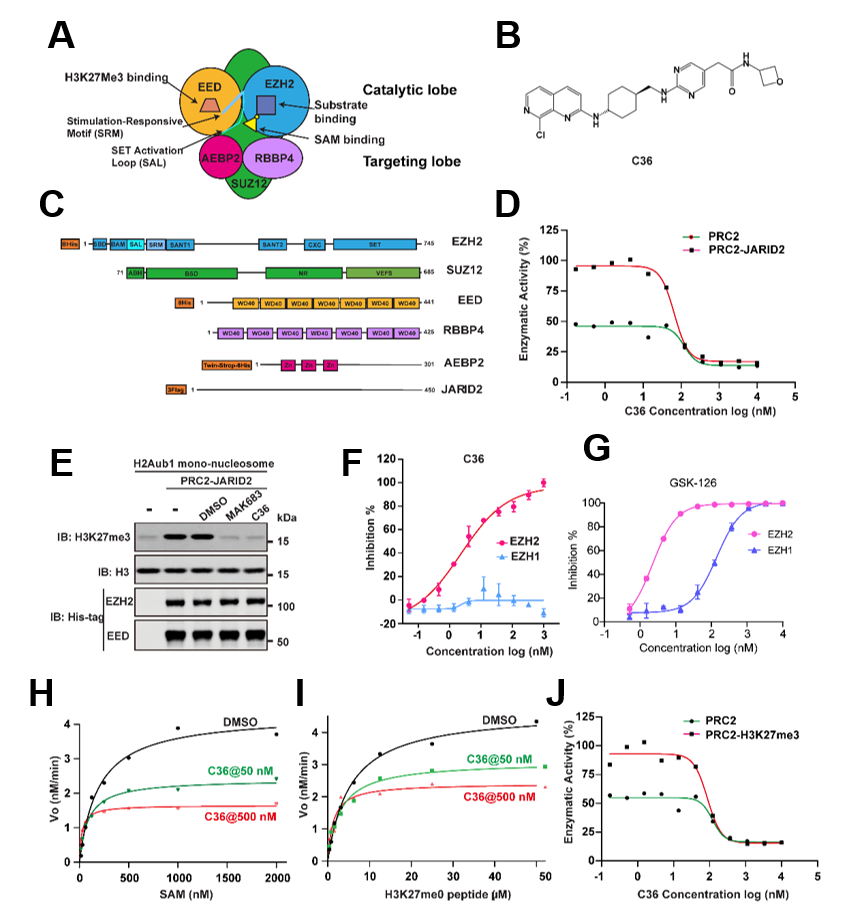
图1. C36是一种独特且高选择性的EZH2-PRC2抑制剂
低骨髓毒性+免疫重塑,具备联合用药潜力
除高选择性外,C36在安全性及联合用药场景中亦展现出明显优势。由于成功规避了EZH1抑制相关的骨髓毒性风险,在小鼠造血集落形成实验中,C36对骨髓祖细胞的毒性分别仅为临床在研药物或者获批上市药物GSK126、EPZ6438和DS-3201的1/13、1/8和1/40,预示其临床耐受性可能大幅改善。
抗肿瘤效力验证显示,C36不仅显著遏制了淋巴瘤细胞的体外增殖及小鼠荷瘤模型的肿瘤进展,还意外发现,C36能够上调I型干扰素通路相关基因(如MHC-I类抗原呈递分子、OAS家族及IFIT家族成员)。深层机制探索表明,C36直接作用于IFNB1基因启动子区域,降低该处H3K27me3富集水平,重启内源性IFNβ转录,进而激活STAT1磷酸化级联并放大干扰素刺激基因的表达。这一独特的免疫重塑效应,使其具备与现行免疫疗法联用的潜力。在体外肿瘤-免疫细胞共杀伤模型中,经C36预处理的肿瘤细胞对CD19特异性CAR-T细胞的裂解敏感性显著增强;在免疫功能健全的LLC荷瘤小鼠中,C36与抗PD-1抗体的联合方案较各单药组显示出更优的抑瘤效果。
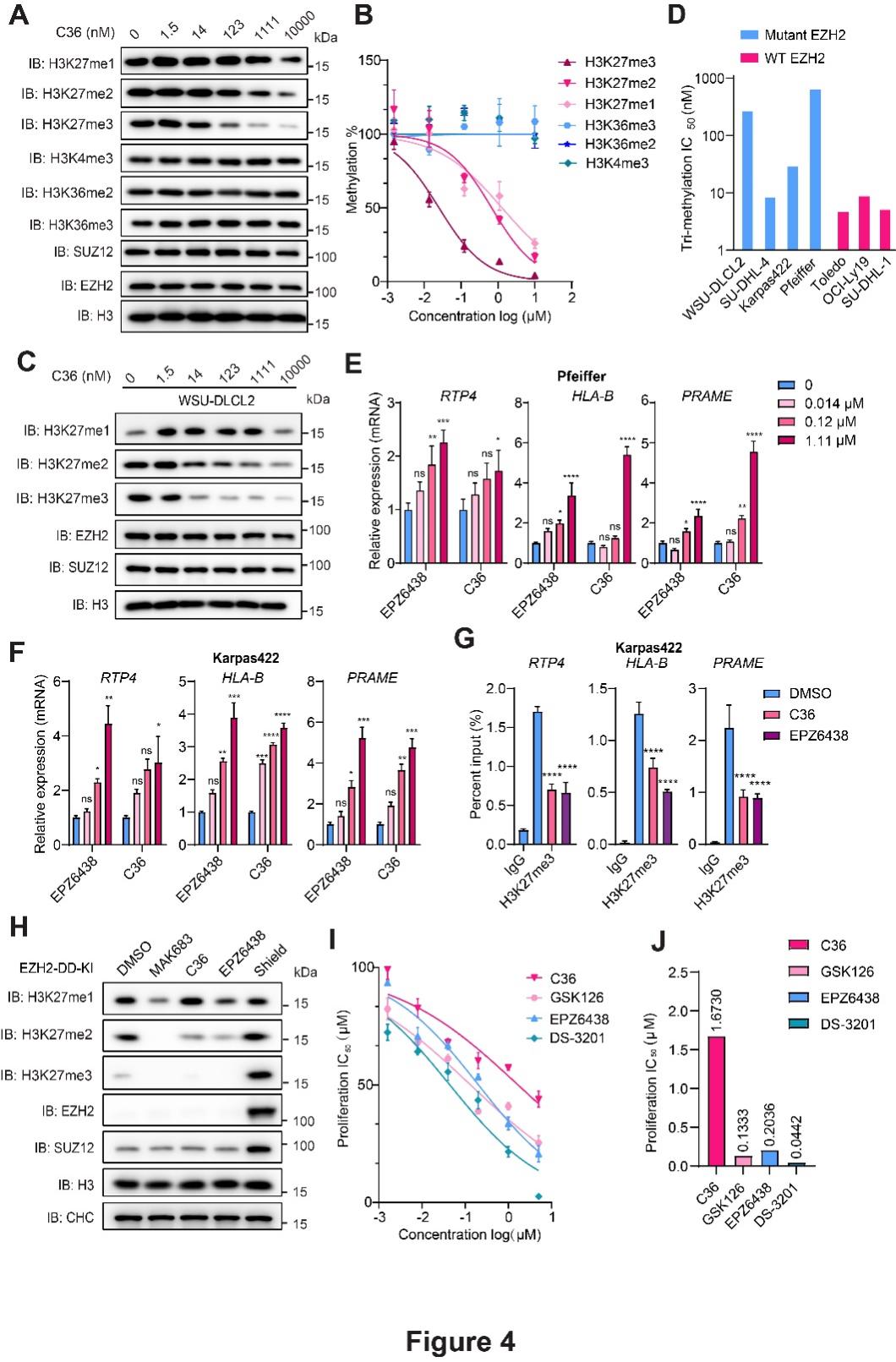
图2. C36在细胞细胞内抑制PRC2
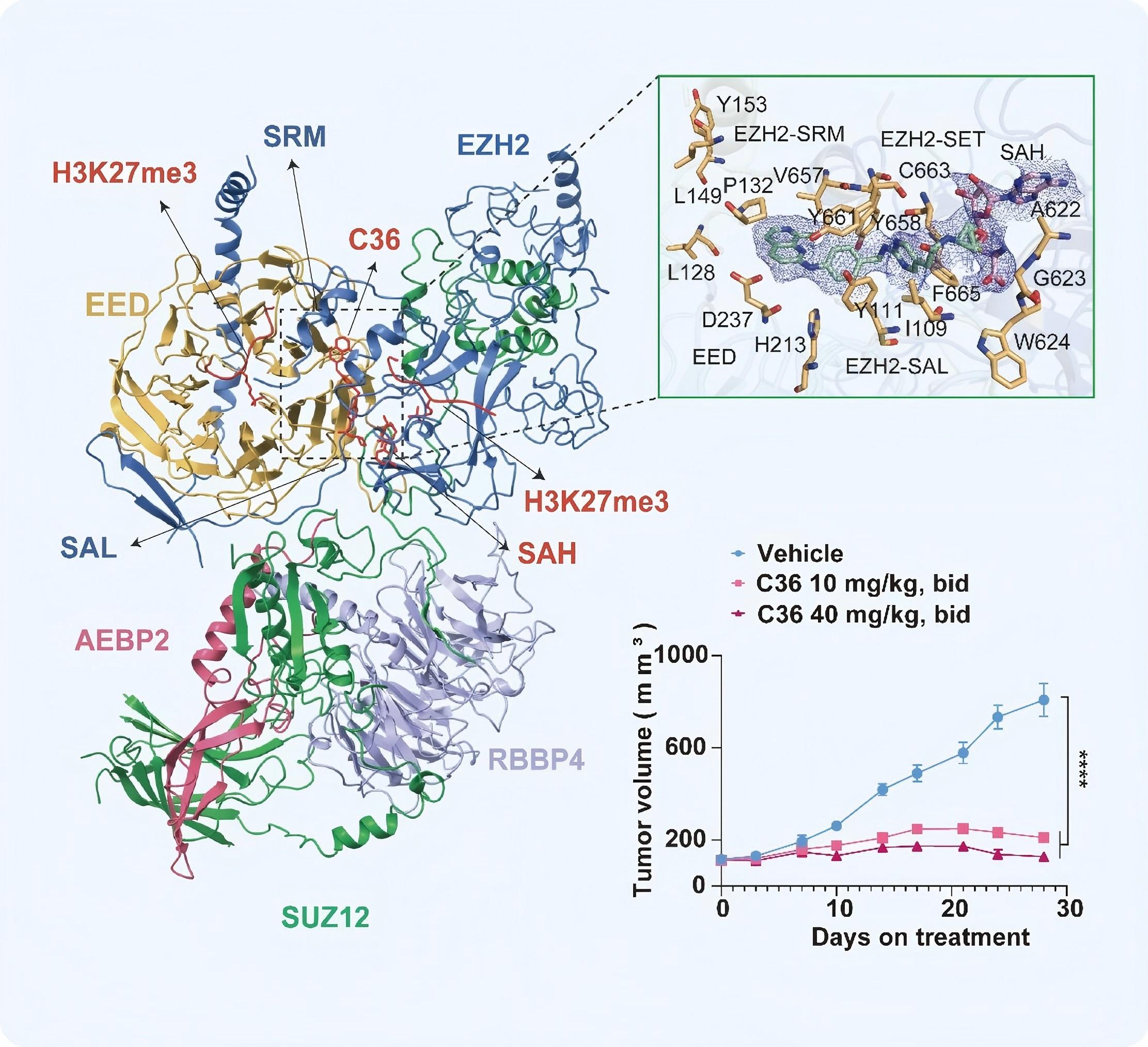
图3.C36结合模型与应用
综上所述,C36凭借其从未被报道的别构结合模式、较高的EZH2/EZH1选择性、极低的毒性以及内在的免疫重塑能力,不仅为解析PRC2复合物的动态调控网络提供了一把高精度分子探针,更为后续设计兼具低毒性与免疫协同效益的表观遗传靶向药物奠定了坚实的临床前基础。
论文链接:https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.76025
 沪公网安备 31011502006855号
沪公网安备 31011502006855号